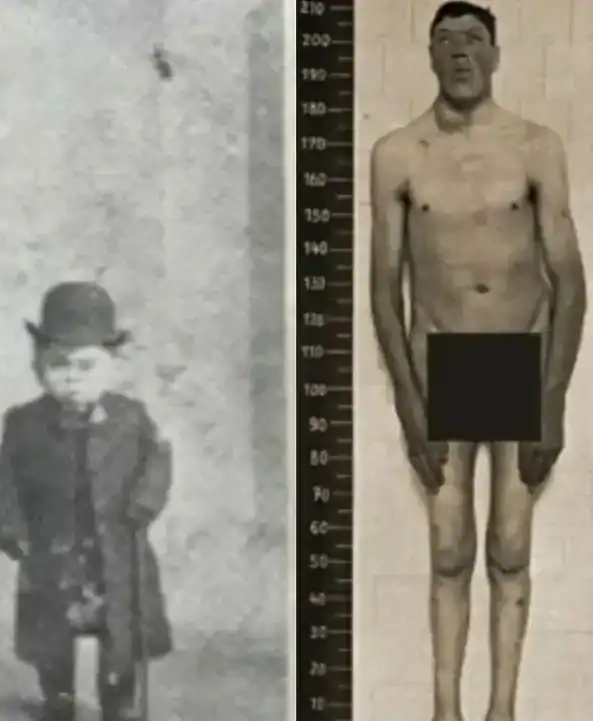





El austriaco Adam Rainer entró en la historia de la medicina por un motivo considerado único hasta hoy. Fue la única persona registrada que desarrolló nanismo y, más tarde, gigantismo a lo largo de su vida. Su caso sigue siendo objeto de estudio en revistas especializadas y congresos de endocrinología.
Nacido en Graz, Austria, en 1899, Rainer pasó gran parte de su juventud siendo considerado extremadamente bajo. A los 18 años medía alrededor de 1,30 metros y fue rechazado para servir en el ejército durante la Primera Guerra Mundial por ser “demasiado pequeño”. Estas restricciones evidenciaban los umbrales de reclutamiento militar de la época y reflejaban una visión rígida de los estándares físicos.
El nanismo que presentaba en su adolescencia no respondía a los síndromes genéticos más comunes, sino a un trastorno del desarrollo aún en estudio. El término nanismo engloba diversas condiciones caracterizadas por una estatura anormalmente baja, que en su caso se manifestaba con extremidades y rasgos faciales proporcionales, pero con una altura total muy reducida.
Todo cambió cuando Adam cumplió 21 años. Inesperadamente, comenzó a experimentar un crecimiento acelerado. En el transcurso de una década superó los dos metros de altura y alcanzó aproximadamente 2,18 metros. Este incremento repentino sorprendió a los médicos de la época y marcó el comienzo de un cuadro clínico opuesto al inicialmente diagnosticado.
El gigantismo observado en Rainer se distingue del nanismo en que implica una producción excesiva de la hormona del crecimiento antes del cierre de las epífisis óseas. Mientras que en el nanismo existen epífisis activas con escasa secreción hormonal, en el gigantismo la glándula pituitaria se vuelve hiperactiva, provocando un alargamiento continuo de huesos y tejidos blandos.
Poco después de detectarse esta aceleración anómala, los especialistas diagnosticaron acromegalia, una condición causada por un adenoma hipofisario. La glándula pituitaria, situada en la base del cráneo, comienza a secretar hormona del crecimiento de manera incontrolada. Con el tiempo, la acromegalia produce deformaciones óseas, engrosamiento de manos y pies, y anomalías faciales.
A medida que la enfermedad avanzó, Rainer sufrió complicaciones graves. Presentó deformidades severas en la columna vertebral, debido a la curvatura forzada de los huesos; asimismo, desarrolló pérdida parcial de la visión, provocada por la presión del tumor sobre el quiasma óptico. En sus últimos años de vida permaneció postrado en cama, con movilidad reducida y dolor crónico.
Para tratar de frenar el crecimiento exagerado se practicó una cirugía de cráneo para extirpar el adenoma responsable. Aunque la intervención logró desacelerar la secreción hormonal, los daños ya eran en gran parte irreversibles: alteraciones óseas y presión sobre tejidos vitales habían dejado secuelas permanentes.
En comparación, Robert Wadlow, registrado como el hombre más alto de la historia, alcanzó 2,72 metros antes de fallecer en 1940 sin provocar nanismo previo. Por otro lado, Khagendra Thapa Magar, conocido por su estatura de 67,8 centímetros antes de su muerte en 2020, ejemplifica el extremo opuesto del crecimiento humano, pero tampoco experimentó una transformación dual como Rainer.
Adam Rainer falleció en 1950, a los 51 años, con aproximadamente 2,40 metros de altura. Desde entonces, ningún otro caso ha reunido las dos condiciones en un solo individuo y su historia se mantiene como un hito singular en la endocrinología clínica.
Hoy, pacientes con acromegalia o trastornos de la hormona del crecimiento reciben tratamientos farmacológicos más avanzados, como análogos de somatostatina y cirugía mínimamente invasiva. No obstante, el caso de Rainer sigue siendo un referente en la literatura médica, recordando la complejidad del sistema hormonal y la necesidad de diagnóstico precoz.

{kind=link}